

A medida que el virus SARS-CoV-2, surgido en diciembre de 2019 en la ciudad china de Wuhan, iba expandiéndose, los científicos ya sabían lo que debían hacer, dada la experiencia recabada años atrás con las pandemias de SARS (2002) y MERS (2012), ambos “primos cercanos” al actual coronavirus.
Por infobae.com
Siguiendo el patrón de estos “parientes letales”, los expertos entendieron que comprender cómo muta y se propaga el coronavirus sería clave para encontrar formas efectivas de lidiar con su contagiosidad, peligrosidad, velocidad de expansión y sobre todo su inexorable mutación a futuro.
Por eso entendieron que recopilar datos epidemiológicos de todo el mundo, procesarlos y analizarlos es clave para llegar al objetivo de comprender al virus para hallar sus puntos más débiles y destruirlo, poniendo fin a esta nueva pandemia que sigue azotando y a todos los sistemas sanitarios del mundo.
Así, conformaron la base de datos internacional llamada Gisaid (las siglas en inglés de “Iniciativa Global para Compartir Datos sobre Influenza”), que fue creada en enero de 2006 para compartir datos sobre el virus de la influenza y ahora para hacer lo mismo sobre el nuevo coronavirus.
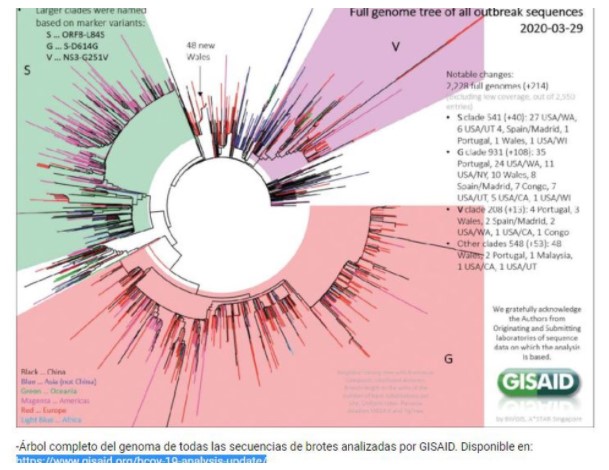
Si bien existen varias bases de datos para las secuencias del genoma, Gisaid es, por mucho, el más popular y grande. Se ha transformado en poco más de un año en “la biblia del coronavirus” por ser el mayor depósito de datos genómicos de los virus de la gripe y ahora del nuevo coronavirus que originó la actual pandemia. En los últimos días, más de 1,2 millones de secuencias del genoma del coronavirus correspondiente a 172 países se han compartido ahora en una popular plataforma de datos en línea, lo que es un testimonio del arduo trabajo de los investigadores de todo el mundo durante la pandemia. Los datos de secuencia han sido cruciales para los científicos que estudian los orígenes del SARS-CoV-2, la epidemiología de los brotes de COVID-19 y el movimiento de variantes virales en todo el planeta.
“Se trata de un logro muy grande en relación al entendimiento de la pandemia ocasionada por el SARS-CoV-2. La plataforma fue creada para compartir datos sobre el virus de la influenza o gripe. Y lo interesante es que, aunque no es la única base de datos de este tipo, sí es la más utilizada para cargar y depositar datos genómicos del SARS-CoV-2”, explicó a Infobae el biólogo y doctor en ciencias Federico Prada, que actualmente es Decano de la Facultad de Ingeniería y Ciencias Exactas de la UADE.
¿Para qué se acumulan este tipo de secuencias?
“El objetivo es entender el origen el coronavirus, saber más de la epidemiología de los brotes y por último tener bien claro cuáles son las variantes que se están moviendo en el mundo. De manera secundaria, todos estos datos nos pueden dar una excelente información acerca de la eficacia de las vacunas para establecer realmente en qué regiones están funcionando y al mismo tiempo comprender de qué manera se está esparciendo el virus por los distintos países del mundo”, analizó el especialista.
Y agregó: “Lo más interesante de todo esto es el momento es lo que se está viviendo a nivel científico y más precisamente a nivel genómico, que es muy similar a lo que ocurrió con la lectura del genoma humano. En primer lugar, al genoma humano tuvimos que leerlo. Esa era la era genómica que concluye con su publicación completa en el 2003. Esa primera etapa tenía un objetivo que era el poder leerlo, secuenciarlo, es decir, tener la información. Luego vino una segunda etapa que tiene que ver con extender la lectura o secuenciación del genoma humano en muchos individuos para comprender las variaciones presentes en la población, que es lo que está pasando ahora con la posibilidad de escalar la secuenciación del genoma del SARS-CoV-2 de manera de tener lo más claro posible sus variantes presentes a nivel mundial. Luego, vino la tercera etapa que llamamos la era pos genómica. Allí se pretende hacer hablar al genoma, que significa transformar todos esos datos en información que nos lleva biológicamente a la función de todos los componentes del genoma del virus que se traducen en proteínas que nos pueden dar una idea acabada de cómo prevenir una terapia para COVID-19”.
Sebastian Maurer-Stroh, que trabaja en Singapur y es asesor científico en GISAID, explicó que “debido a que los países envían datos de tantas partes del mundo, existe un sistema en el que podemos observar cómo se propaga el virus por el mundo y ver si las medidas de control y las vacunas aún funcionan”.
Cuando COVID-19 comenzó a propagarse en China el equipo de GISAID se acercó de inmediato a investigadores y políticos de todo el mundo para comprender qué barreras podrían evitar que compartan datos genómicos sobre el SARS-CoV-2.

“Por ejemplo, cuando los investigadores de África Occidental dijeron que carecían de formación en bioinformática, un científico afiliado a GISAID en Senegal comenzó a realizar talleres sobre secuenciación, análisis y cómo utilizar las herramientas en la plataforma. Algunas de las características de GISAID permiten a los investigadores ver cómo los genomas que se han registrados se relacionan con otros, o explorar dónde aparecen nuevas variantes día a día”, aclaró Maurer-Stroh, que dice que la popularidad del sitio se debe principalmente a su mecanismo de intercambio y la calidad de sus herramientas para la visualización y el análisis de secuencias.
Algunos países ricos han subido una gran cantidad de secuencias y representan la mayor parte de sus regiones. Por ejemplo, al 20 de abril, Estados Unidos había compartido 303,359 secuencias y el recuento del Reino Unido era de 379,510 secuencias. Argentina sumó más de 2300 secuencias en un año.
Para buscar o descargar secuencias de GISAID, o utilizar las herramientas de análisis genómico de la plataforma, las personas deben registrarse con su nombre y aceptar términos que incluyen no publicar estudios basados en los datos sin reconocer a los científicos que cargaron las secuencias, e incluso contactarlos. para preguntar sobre la colaboración. Este control de acceso ha molestado a algunos científicos, que argumentan que no debería haber barreras que se interpongan en el camino del acceso.
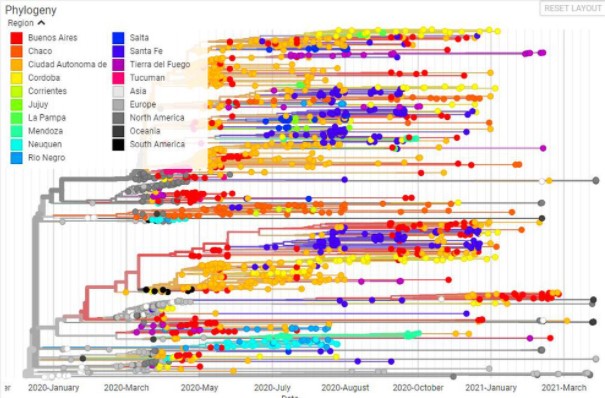
Filogenia del coronavirus en Argentina
Pero GISAID probablemente “no habría alcanzado la marca del millón sin ese enfoque, porque no habría tenido garantías contra la explotación”, especula Tulio de Oliveira, director de la Plataforma de Secuencia e Innovación en Investigación KwaZulu-Natal en Durban, Sudáfrica. Él dice: “Esta es la primera vez que veo a personas compartiendo tantos datos antes de la publicación”.
Las políticas de GISAID para la publicación de datos rápida y completa se basan en las establecidas para los proyectos de recursos comunitarios. Estas políticas han sido empleadas con éxito anteriormente, por ejemplo, por el Proyecto Internacional HapMap , un proyecto para mapear, y hacer disponible gratuitamente, datos sobre variaciones de secuencia de ADN en el genoma humano.
El consorcio GISAID estará integrado por científicos de todo el mundo que trabajan en los campos de la virología animal y humana, la epidemiología y la bioinformática, así como expertos en cuestiones de propiedad intelectual. Se formará un panel internacional de científicos distinguidos para gobernar la carta y asesorar al consorcio. Como esfuerzo de colaboración internacional, GISAID ofrece muchos beneficios al mundo en su conjunto, así como a los científicos individuales y a los grupos que participan en el consorcio.
“Toda esta información que se está almacenando en este repositorio es clave a futuro. Nosotros tenemos dos horizontes a alcanzar. Primero, integrar todos los datos genéticos del virus con los del humano. Hay que cruzar esa información de manera tal de abordar una nueva tecnología “Hi Through” que es el paradigma de la fenómica, que hace referencia al fenotipo de una persona , ya sea una enfermedad interna, como un tipo de cáncer, o infecciosa como la que ocurre con el coronavirus. Y luego, esta manifestación que se da en el humano o fenotipo como respuesta a esta agresión, hay que cruzarla con los datos del ambiente para terminar de pulir el fenotipo de las personas”, sostuvo Prada.
Y concluyó: “Por eso es clave integrar la información existente con la genómica o perfil genotípico del humano e incorporar la fenómica o manifestaciones clínicas de las personas infectadas en función del ambiente”.
Antes del nacimiento de este sitio, muchos países retenían la información genómica por diversas razones. Un temor era que los países que generaban los datos no obtendrían crédito o no obtendrían los beneficios de la investigación derivada de su trabajo original de secuenciación. Pero después de dos años de negociaciones entre gobiernos y científicos sobre acuerdos de intercambio de datos, se lanzó GISAID.
Epidemiología genómica del SARS-CoV-2

El genoma de cualquier organismo son las instrucciones genéticas necesarias para su funcionamiento y reproducción. En el caso del coronavirus las instrucciones están codificadas en 30 mil letras de ARN (A, C, G y U). Cuando una célula es infectada por el coronavirus, se reproducen millones de copias del genoma original. A medida que la célula copia ese genoma, a veces se generan “errores” que consisten en la variación de una letra (puede, por ejemplo, cambiar la U por la G). Esos errores tipográficos que se producen al azar son conocidos como mutaciones y se van acumulando a medida que el virus se propaga. Pero no todas las mutaciones dan origen a una nueva cepa. Se habla de una nueva cepa cuando las modificaciones (mutaciones) generan cambios relevantes en la capacidad del virus de generar la enfermedad, en su estabilidad o en la capacidad de virus de ser bloqueado por el sistema inmunológico.
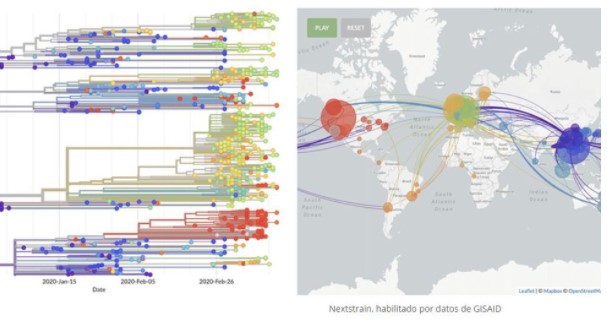
Durante la replicación y transmisión, los genomas virales acumulan mutaciones que pueden usarse para definir distintas variantes. La secuenciación del genoma completo puede identificar estas mutaciones y rastrear la transmisión de variantes entre individuos, poblaciones y países de todo el mundo. Este enfoque de epidemiología genómica se utilizó para monitorear brotes anteriores de infección por el virus del Ébola y el virus del Zika y se ha utilizado ampliamente para reconstruir la propagación del SARS-CoV-2. Una de estas cepas de SARS-CoV-2, denominada ‘B.1’, se transmitió inicialmente a Italia, donde provocó un brote en Lombardía antes de seguir circulando por Europa y en los Estados Unidos, donde sembró un brote en la ciudad de Nueva York. Esta cadena de transmisión en todo el mundo se reconstruyó mediante el seguimiento de cuatro mutaciones en la cepa SARS-CoV-2 B.1. El genoma viral del SARS-CoV-2 se diversificó rápidamente en diferentes cepas tras la transmisión zoonótica inicial del virus ancestral.
Durante su difusión inicial, los laboratorios de investigación de todo el mundo secuenciaron rápidamente miles de genomas virales y se compartieron en bases de datos de acceso abierto, como la base de datos EpiCoV de GISAID y el conjunto de datos Our World in Data COVID-19 . Estas secuencias, junto con los metadatos (incluida la ubicación, la fecha y el método de muestreo) y herramientas como Nextstrain , podrían usarse para rastrear la propagación de las cepas del SARS-CoV-2 A pesar de la utilidad de la epidemiología genómica, los datos deben interpretarse con cautela. Aunque la infección con diferentes cepas puede descartar una cadena de transmisión entre dos individuos, las infecciones por la misma cepa no necesariamente demuestran un vínculo directo en la cadena de transmisión.